






应用领域
技术路线

分析内容
|
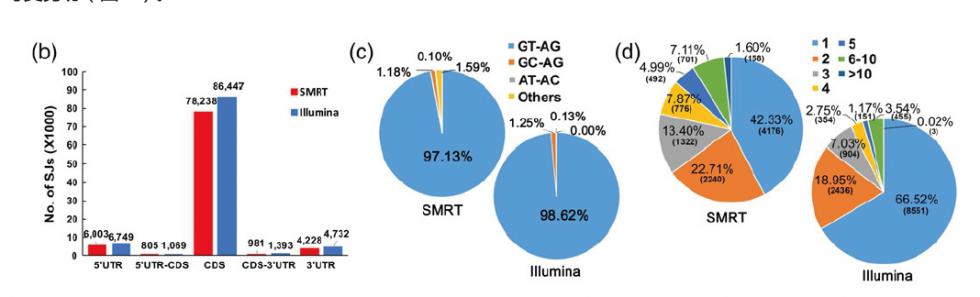
1. 标准信息分析 a) 原始测序数据统计及质控 b) Reads分类 c) Reads聚类和校正 d) 全长isoform数据统计 e) 比对参考基因组 f) 新转录本预测 g) 新转录本功能注释 1) Nr注释 2) GO功能注释 3) KEGG代谢通路注释 4) SwissProt蛋白注释 |
h) 基因结构分析
1) lncRNA分析
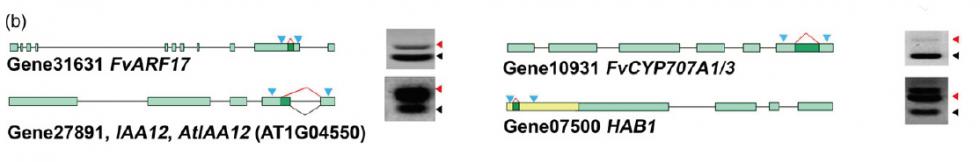
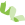
2) 可变剪切分析
3) 可变多聚腺苷酸化分析
4) 融合基因分析
5) SNP/InDel分析
6) 开放阅读框分析
7) 转录因子分析
8) 基因结构优化
2. 定制化信息分析 a) 二代数据校正三代数据(需有Illumina数据) b) 基因定量及差异表达分析(需有Illumina数据) c) 多组学关联分析(如甲基化、蛋白组、miRNA) |