






应用领域
技术路线

分析内容
|
1. 标准信息分析
1.1原始测序数据统计及质控
1.2Reads分类
1.3 Reads聚类和校正
1.4全长isoform数据统计
1.5 基因基本功能注释
a) Nr注释
b) GO功能注释
c) COG/KOG注释
d) KEGG代谢通路注释
e) SwissProt蛋白注释
1.6基因高级功能注释
a) 预测编码蛋白框(CDS)
b) 转录因子分析
c) R基因分析(植物)
d) 蛋白结构域分析(Pfam, SMART)
e) TMHMM跨膜螺旋结构预测
|
f) SignalP信号肽结构预测
g) 蛋白O-GlcNAc糖基化位点预测(哺乳动物)
h) ProP弗林蛋白酶裂解位点预测(真核生物)
1.7结构分析
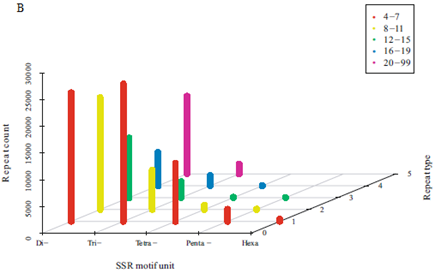
a) 串联重复单元检测(SSR)
b) lncRNA分析
c) 可变剪切分析
2. 定制化信息分析
a) 二代数据校正三代数据(需有Illumina数据)
b) 基因定量及差异表达分析(需有Illumina数据)
c) 多组学关联分析(如甲基化、蛋白组、miRNA)
|
拟穴青蟹三代全长转录组测序
研究样本
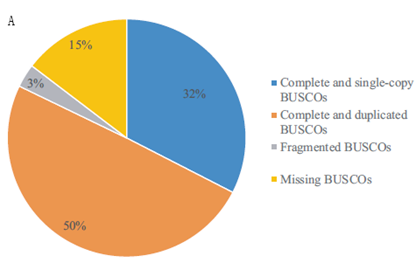
研究结果